
Here is a detailed description of how the example dataset was generated from the original chromatograms accessible from the main page. Please note, however, that the method outlined here is only one among many others; other paths may be more suitable to your data.
First, the two traces were imported into a contig editor (namely, Sequencher) and assembled using the following assembly parameters:
Here is how the aligned chromatograms look like:
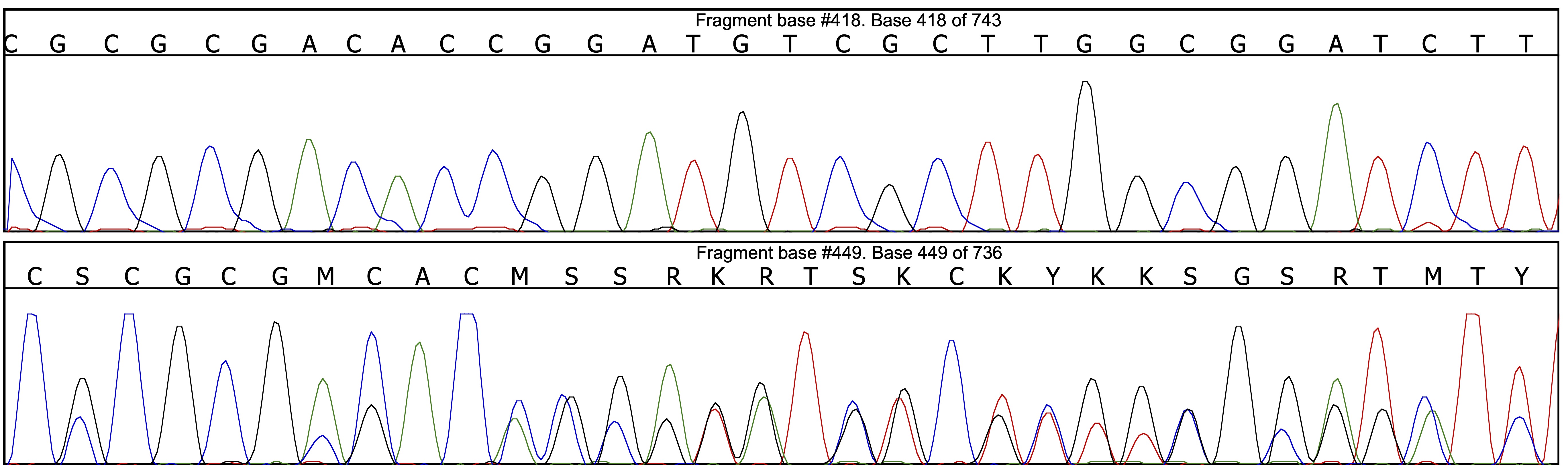
Many double peaks are not properly called, and it is always a good idea to start by correcting as many calling mistakes as possible before submitting the sequences to Champuru; this can save a lot of time in the following steps. A convenient and fast way to do so is to is by looking for positions where the forward and reverse chromatograms are incompatible in the alignment (represented by black dots on the figure below):
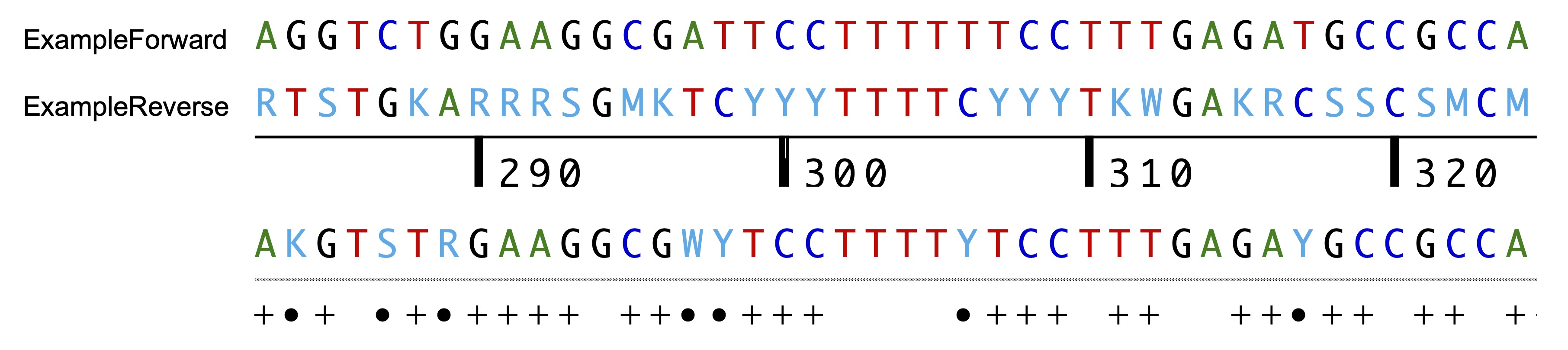
Once all base-calling mistakes visible as sequence incompatibilities are corrected in the first alignment position, one may want to find the second, alternative alignment in order to correct other mistakes. This can be achieved onscreen be dragging one sequence relatively to the other until finding another alignment that minimizes the number of incompatible positions. The distance observed at the end of each chromatogram between the two end peaks can also help to estimate the length difference between the two haplotypes and how far away one chromatogram needs to be dragged relative to the other in order to find the alternative alignment position:
Here the distance between the two end peaks shows unambiguously that the length difference between the two haplotype is 2 bp; hence, the second alternative alignment can be obtained from the first one by dragging one chromatogram 2 bp in one direction or the other relative to the other chromatogram.
The same information can also be obtained by submitting the partially corrected sequences to Champuru. The output will look like:
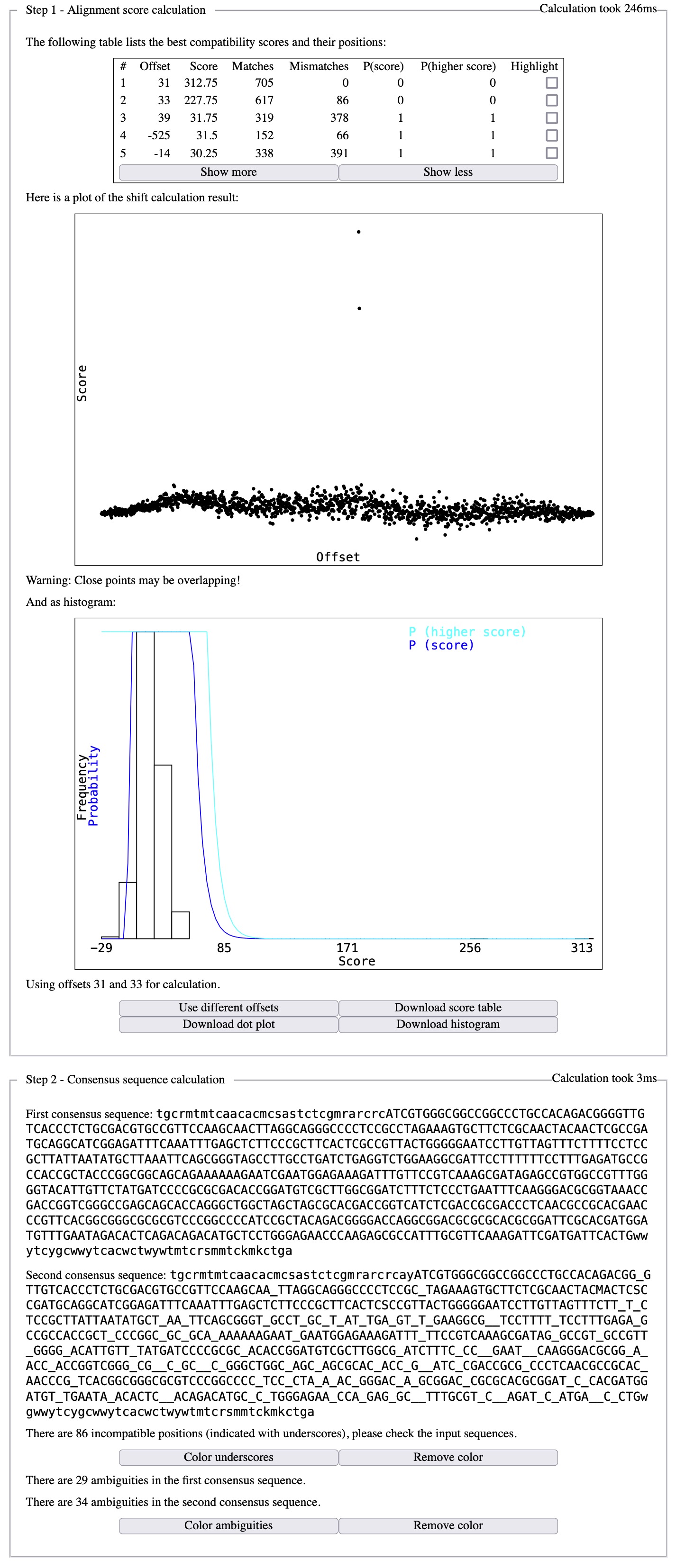
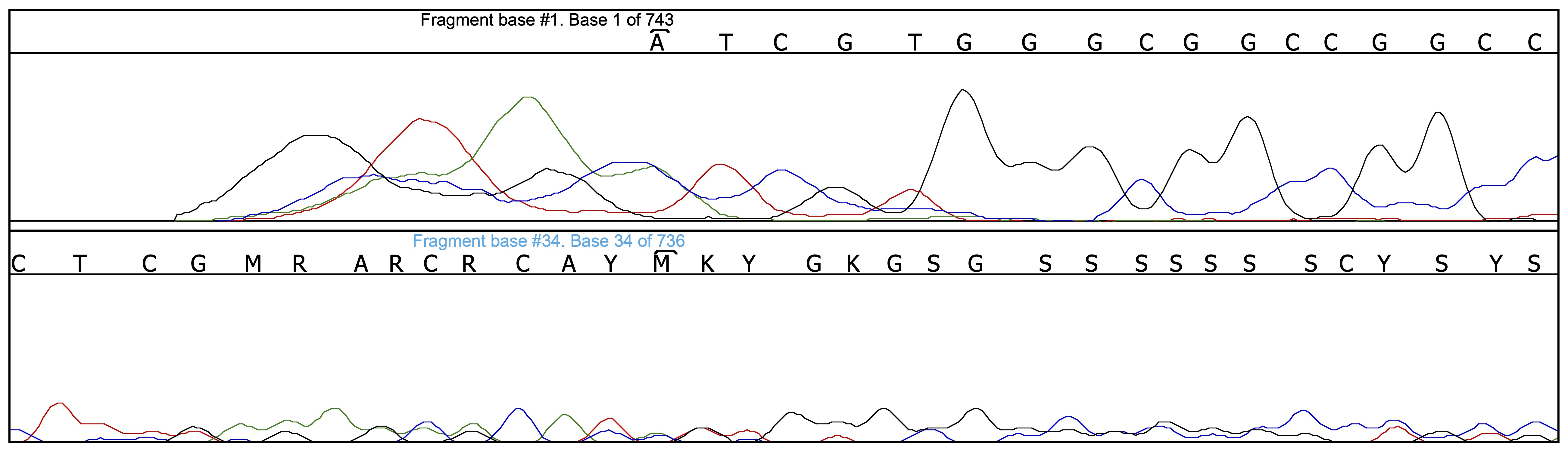
The offset between the forward and reverse chromatogram for the first aligment (best compatibility score) being 31 bp, and 33 bp for the second alignment (second best compatibility score), one alignment can be obtained from the other by dragging one chromatogram by 33 - 31 = 2 bp. Alternatively, one can use the offset information to position directly the chromatograms correctly in front of each other: here is for the first alignment (offset = 31 bp)
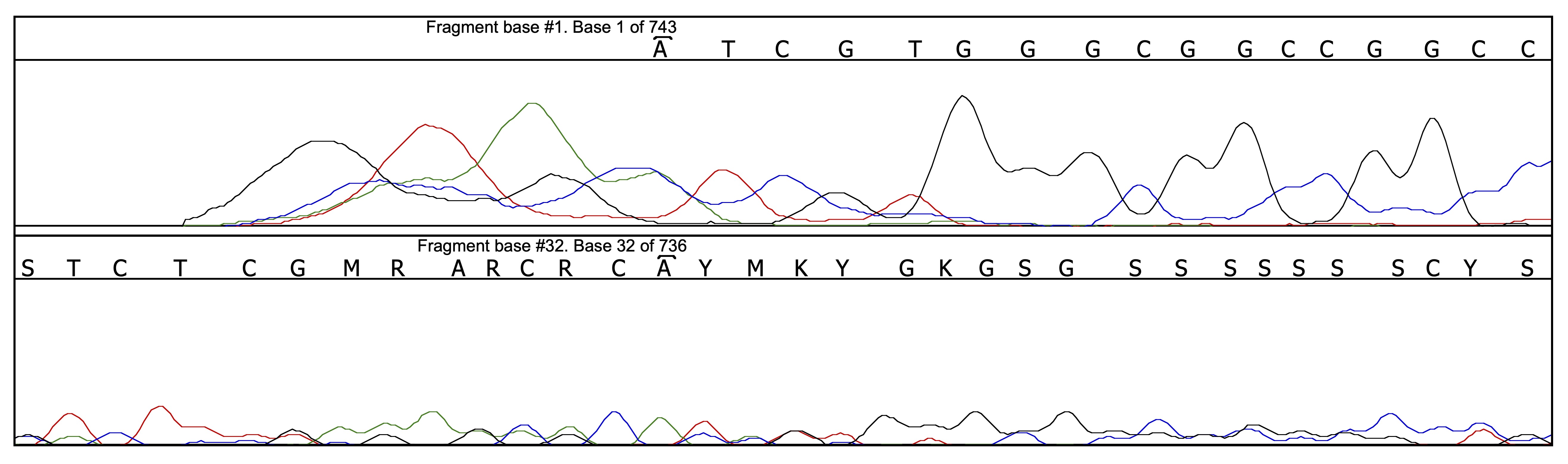
and here is for the second (offset = 33 bp).
Once all easily detected mistakes have been corrected, it is now time to submit the corrected sequences to Champuru; the output will look like:
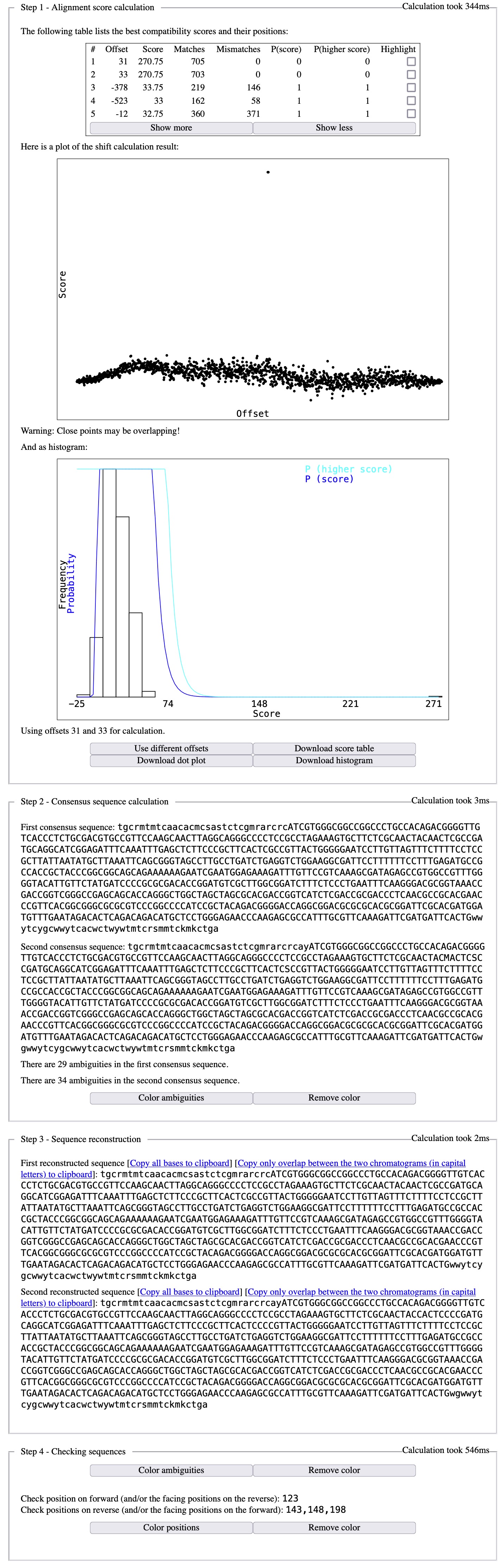
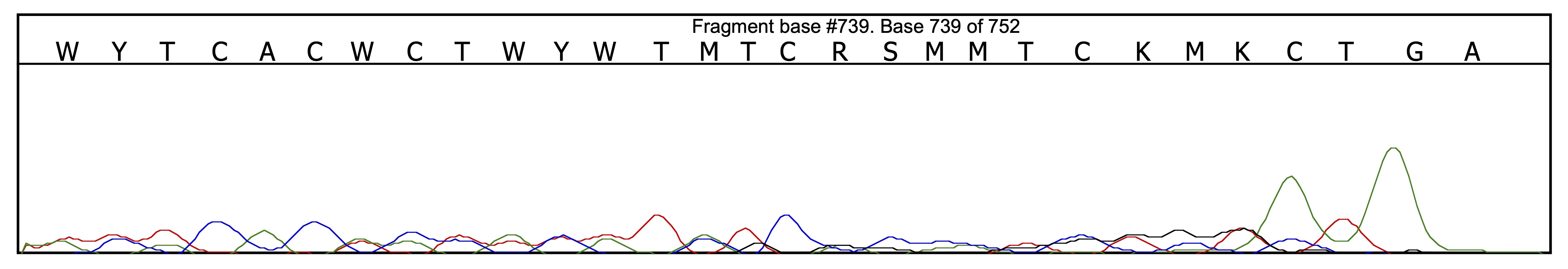
Thanks to all the pre-cleaning of base-calling mistakes, there are only 4 problems left to be corrected, one in the forward chromatogram and the other three in the reverse chromatogram. Let us have a look, for instance, at position 143 on the reverse chromatogram:
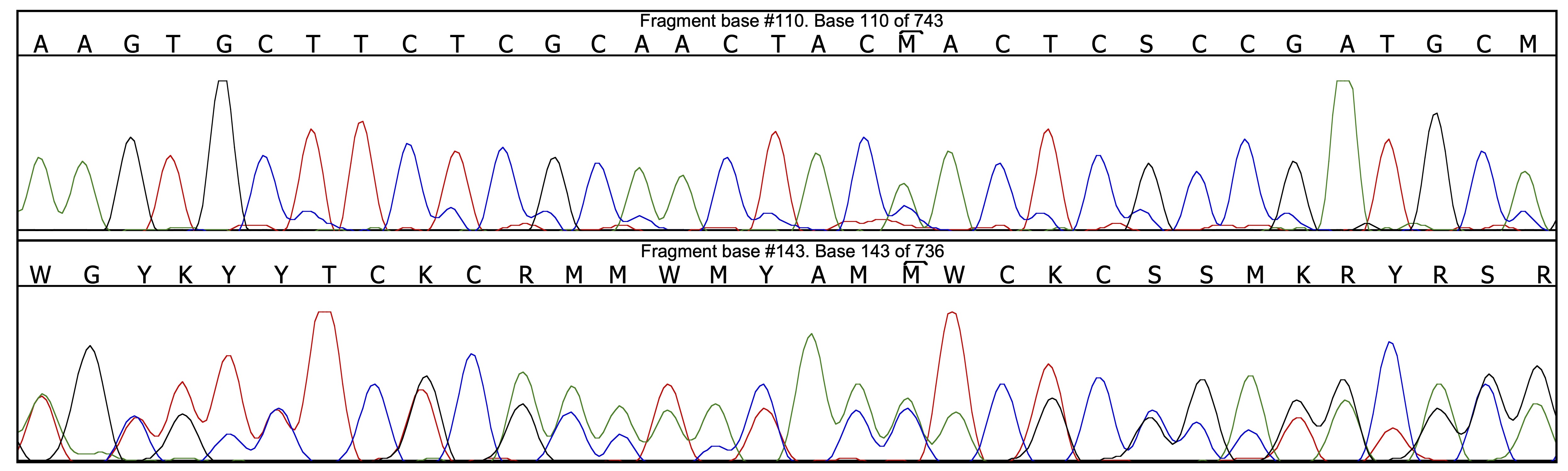
One may be suprised to see that the base calling on the reverse chromatogram at that position is perfectly correct; actually, the problem is located on the other chromatogram (M instead of A) due to the presence of an artefactual peak. The locations given by Champuru for the problems it detects are only approximate, and one sometimes needs to look closely at the surroundings of the indicated location in order to find out what the problem is.
After the last remaining problems are corrected, Champuru is run one last time on the corrected chromatogram sequences in order to produce the reconstructed haplotype sequences. The corrected chromatogram sequences are the one provided on the main page as example dataset.
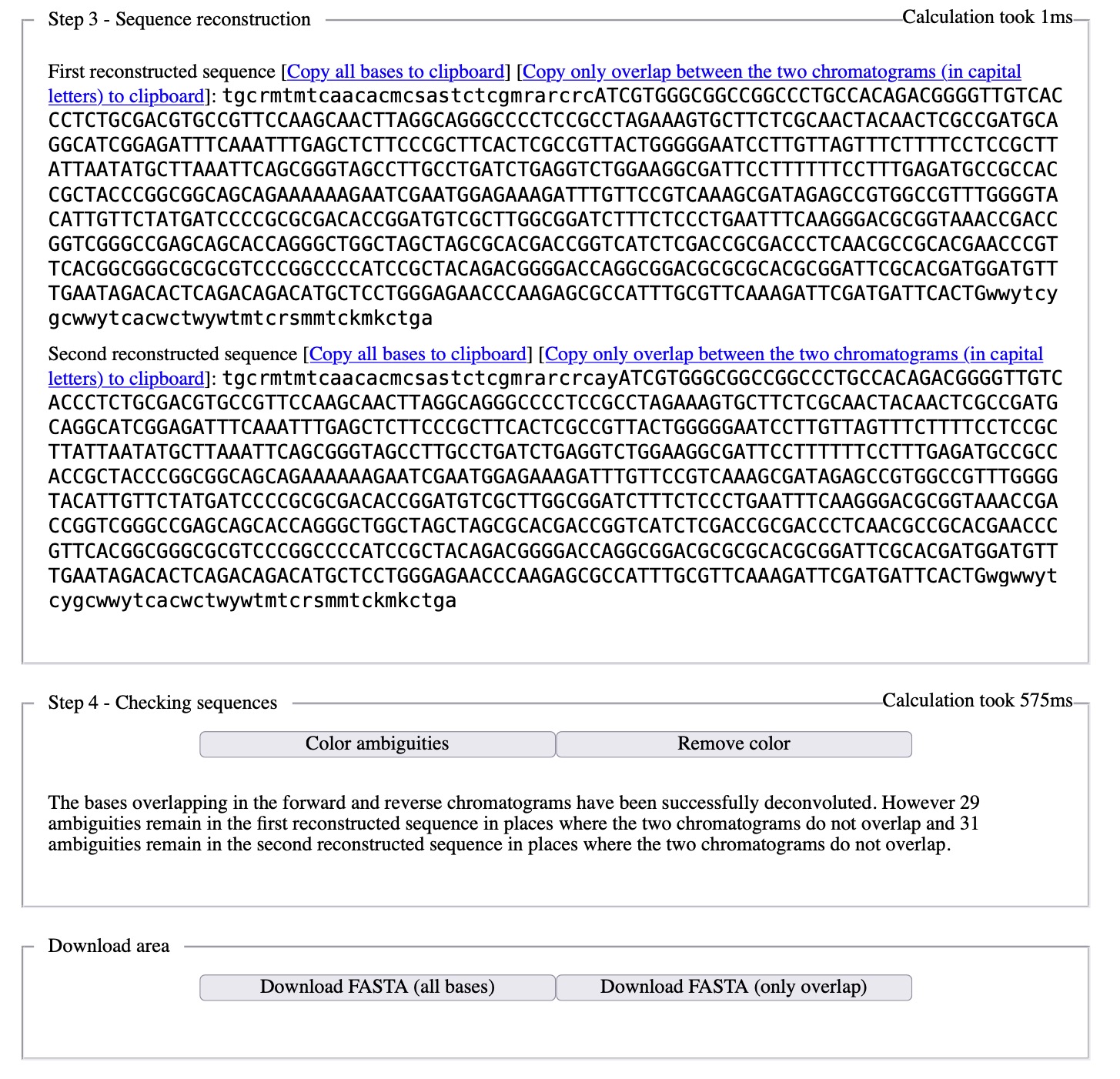
The haplotype sequences can finally be saved on disk in a single FASTA file, or copy/pasted into other applications.
This detailed description of the process may look complicated, but with a bit of training it it possible to clean a pair of chromatogram and obtain the corresponding haplotype sequences in less than five minutes. Have a try!